Understanding Carbamoyl-Phosphate Synthetase 1 Deficiency: A Comprehensive Guide
Carbamoyl-Phosphate Synthetase 1 Deficiency (CPS1 Deficiency) is a rare genetic metabolic disorder affecting the urea cycle. This crucial cycle is responsible for removing ammonia, a toxic waste product, from the body. When CPS1, the first enzyme in the urea cycle, is deficient or absent, ammonia builds up in the blood, leading to hyperammonemia. This condition can cause severe neurological damage and, if left untreated, can be life-threatening. Early diagnosis and management are critical for improving the outcomes for individuals with Carbamoyl-Phosphate Synthetase 1 Deficiency.
What is Carbamoyl-Phosphate Synthetase 1?
Carbamoyl-Phosphate Synthetase 1 (CPS1) is an enzyme located in the mitochondria of liver cells. It plays a vital role in the urea cycle by catalyzing the first committed step: the synthesis of carbamoyl phosphate from ammonia, bicarbonate, and ATP. Without sufficient functional CPS1, the urea cycle cannot effectively convert ammonia into urea, which is then excreted in urine. This leads to the accumulation of ammonia in the bloodstream, causing hyperammonemia.
Causes and Genetics of CPS1 Deficiency
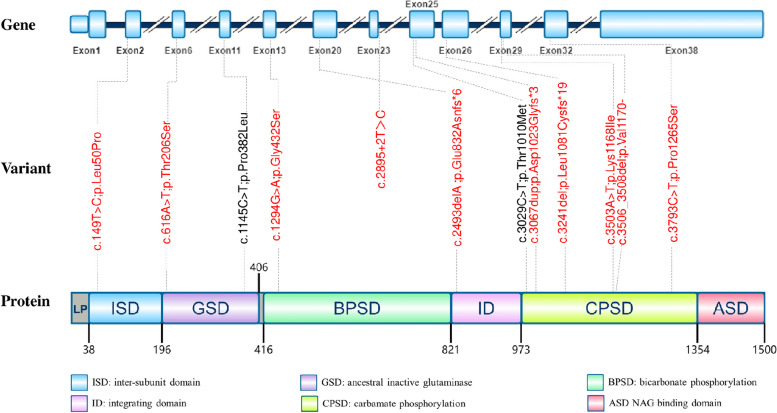
Carbamoyl-Phosphate Synthetase 1 Deficiency is caused by mutations in the CPS1 gene. This gene provides the instructions for making the CPS1 enzyme. The disorder is inherited in an autosomal recessive manner, meaning that both parents must carry a copy of the mutated gene for their child to be affected. Individuals who carry only one copy of the mutated gene are carriers and typically do not exhibit symptoms of the deficiency.
Genetic testing can confirm the diagnosis of Carbamoyl-Phosphate Synthetase 1 Deficiency and can also be used for carrier screening and prenatal diagnosis.
Symptoms of Carbamoyl-Phosphate Synthetase 1 Deficiency
The symptoms of Carbamoyl-Phosphate Synthetase 1 Deficiency can vary depending on the severity of the enzyme deficiency and the age of onset. There are generally two main forms:
- Neonatal-onset: This is the most severe form and typically presents within the first few days of life. Symptoms include lethargy, poor feeding, vomiting, irritability, seizures, and coma. Without prompt treatment, this form is often fatal.
- Late-onset: This form can present in infancy, childhood, or even adulthood. Symptoms may be milder and more intermittent, including episodes of vomiting, confusion, ataxia (lack of coordination), and behavioral changes. These episodes are often triggered by illness, stress, or high-protein intake.
Hyperammonemia, regardless of the age of onset, can lead to irreversible brain damage, developmental delays, and intellectual disability. Therefore, recognizing the symptoms and seeking timely medical attention are crucial.
Diagnosis of CPS1 Deficiency
The diagnosis of Carbamoyl-Phosphate Synthetase 1 Deficiency typically involves a combination of clinical evaluation, laboratory testing, and genetic analysis.
- Ammonia levels: Elevated ammonia levels in the blood are a key indicator of urea cycle disorders, including Carbamoyl-Phosphate Synthetase 1 Deficiency.
- Amino acid analysis: Blood and urine amino acid analysis can reveal characteristic patterns associated with CPS1 deficiency, such as low levels of citrulline.
- Urine orotic acid: Urine orotic acid levels are usually normal, which helps differentiate CPS1 deficiency from other urea cycle disorders like ornithine transcarbamylase (OTC) deficiency.
- Enzyme assay: A liver biopsy can be performed to directly measure the activity of CPS1 enzyme. However, this is an invasive procedure and is not always necessary for diagnosis.
- Genetic testing: Genetic testing of the CPS1 gene can confirm the diagnosis by identifying disease-causing mutations. It can also be used for carrier screening and prenatal diagnosis.
Treatment and Management
The primary goal of treatment for Carbamoyl-Phosphate Synthetase 1 Deficiency is to lower ammonia levels and prevent hyperammonemic crises. This typically involves a combination of dietary management, medications, and, in some cases, liver transplantation.
Dietary Management
A low-protein diet is essential to reduce the production of ammonia from protein breakdown. The amount of protein allowed in the diet varies depending on the individual’s age, weight, and severity of the deficiency. A registered dietitian specializing in metabolic disorders can help develop a personalized dietary plan.
Specialized medical formulas that are low in protein but provide essential amino acids may also be necessary to ensure adequate nutrition.
Medications
- Ammonia scavengers: Medications such as sodium benzoate and sodium phenylbutyrate help to remove ammonia from the bloodstream by providing alternative pathways for nitrogen excretion. These medications are typically taken daily and are essential for long-term management.
- Arginine supplementation: Arginine is an amino acid that is deficient in individuals with Carbamoyl-Phosphate Synthetase 1 Deficiency. Supplementation with arginine can help to improve the function of the urea cycle.
- N-carbamylglutamate (Carbaglu): This medication acts as a CPS1 activator and can help to improve the enzyme’s function in some individuals with CPS1 deficiency.
Liver Transplantation
Liver transplantation is a potentially curative treatment option for severe cases of Carbamoyl-Phosphate Synthetase 1 Deficiency. A successful liver transplant provides a source of functional CPS1 enzyme, allowing the urea cycle to function normally and preventing hyperammonemia. However, liver transplantation is a major surgical procedure with its own risks and complications, and it is not suitable for all individuals.
Living with CPS1 Deficiency
Living with Carbamoyl-Phosphate Synthetase 1 Deficiency requires ongoing medical management and close monitoring. Regular follow-up appointments with a metabolic specialist are essential to monitor ammonia levels, adjust medications, and address any complications. Parents and caregivers play a crucial role in managing the condition, ensuring adherence to dietary restrictions and medications, and recognizing early signs of hyperammonemia.
Support groups and online communities can provide valuable resources and emotional support for individuals and families affected by Carbamoyl-Phosphate Synthetase 1 Deficiency. [See also: Related Article Titles]
Research and Future Directions
Ongoing research is focused on developing new and improved treatments for Carbamoyl-Phosphate Synthetase 1 Deficiency. This includes gene therapy, which aims to correct the underlying genetic defect by delivering a functional copy of the CPS1 gene to liver cells. Other research efforts are focused on developing novel medications that can more effectively lower ammonia levels and prevent hyperammonemic crises.
Conclusion
Carbamoyl-Phosphate Synthetase 1 Deficiency is a serious metabolic disorder that requires lifelong management. Early diagnosis, prompt treatment, and ongoing monitoring are essential to prevent neurological damage and improve the long-term outcomes for affected individuals. With advances in medical care and ongoing research, the outlook for individuals with Carbamoyl-Phosphate Synthetase 1 Deficiency continues to improve. Understanding the complexities of Carbamoyl-Phosphate Synthetase 1 Deficiency allows for better care and support for those affected. The key to successful management of Carbamoyl-Phosphate Synthetase 1 Deficiency involves early detection, consistent treatment and a strong support system. Addressing Carbamoyl-Phosphate Synthetase 1 Deficiency requires a multidisciplinary approach involving medical professionals, dietitians, and support groups. Continuous research into Carbamoyl-Phosphate Synthetase 1 Deficiency offers hope for improved treatments and outcomes. Managing Carbamoyl-Phosphate Synthetase 1 Deficiency effectively can significantly improve the quality of life for affected individuals and their families.

